
UK MHRA: 2024-2025 Roadmap of Future Regulatory Framework

EU Commission: New Dashboard Monitoring Availability of Medical Devices on the EU Market
January 9, 2024
EU Commission Publishes Language Translation Requirements under the MDR & IVDR
January 18, 2024Today the MHRA:
- Updated its webpage “Implementation of the Future Regulations”, and
- Released a new “Roadmap towards the future regulatory framework for medical devices”.
The updates are available: HERE
Implementation of the Future Regulations
This webpage was updated with the following information.
Post-market Surveillance (PMS)
The Post-market Surveillance timeframe was updated.
The previous text stated that the PMS legislation would be laid down winter of 2023 and was expected to apply from mid-2024. It now states that the PMS legislation is expected to apply towards the end of 2024.
Future Regulations
A new section was added, to provide details on the proposed new medical device regulation.
Currently, the UK legislation is based on the old EU Directives (MDD/AIMDD/IVDD). The MHRA intends to lay down new legislation, which will align more closely with the EU MDR/IVDR.
The current timing for the new legislation is:
- First half of 2024 – stakeholder discussions
- End of 2024 – draft legal text for public consultation (published by the World Trade Organization)
- 2025 – laid in Parliament and goes into force
The new legislation will consist of the following changes – including their continued aim to implement a framework for international recognition.
- “Introduce several improvements for implantable medical devices; up-classifying them which will result in more stringent pre and post market requirements and requiring manufacturers to provide implant cards to enable patients to know which device they have had implanted.
- Ensure devices have a unique device identifier (UDI).
- Change the classification of several types of devices, specifically increasing the class of certain software as a medical device and aligning IVD classifications with those of the International Medical Device Regulators Forum.
- Strengthen the requirements for quality management systems and technical documentation.
- Introduce a framework for international recognition, enabling swifter access for devices already approved by comparable regulators as well as for those who have Medical Device Single Audit Program (MDSAP) certificates.
- Include new requirements for exempt in-house manufactured devices and custom-made devices.
- Include new requirements for the claims manufacturers can make about their medical devices requiring them to align with their statement of intended purpose.
- Include new requirements for clinical investigations.
- Bring the essential requirements for medical devices being placed on the market in GB into greater alignment with those of the EU. This will include cybersecurity requirements for software as a medical device including for artificial intelligence.
- Clarify the requirements for conformity assessments and approved bodies.
- Clarify the requirements for economic operators; manufacturers, importers and distributors, and introduce a requirement to have a Person Qualified in Regulatory Compliance.”
Note that the above does not impact the MHRA’s current acceptance of CE Marked devices in Great Britain.
The MHRA has granted a transition period, allowing CE Marked devices through 2028-2030 (depending on type of device). Implementation of the new UK medical device regulation (“UKCA Marking”) is currently not expected to change this.
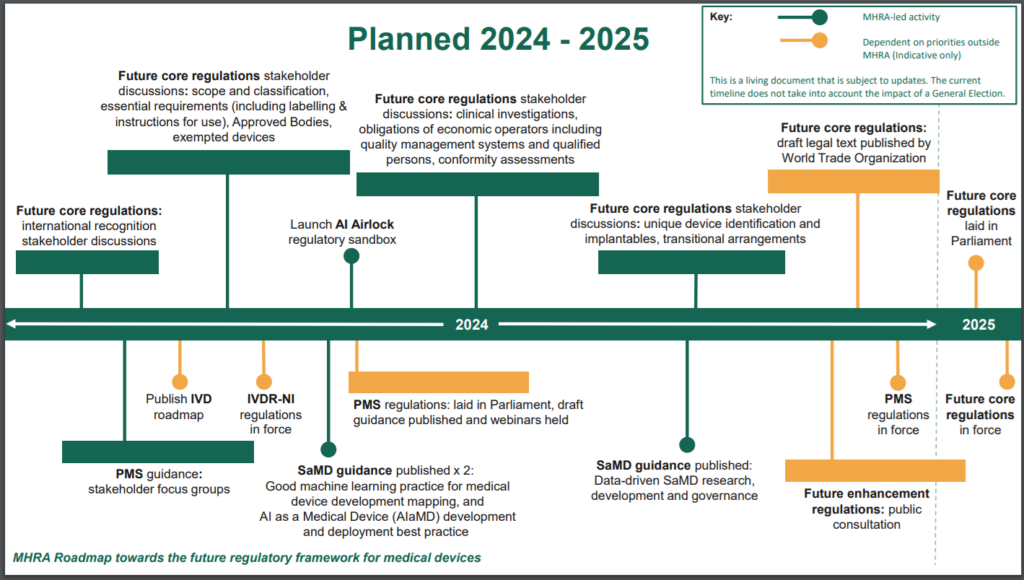
Roadmap on the Future Regulatory Framework
The MHRA published a planned 2024-2025 roadmap.
It outlines the various stakeholder discussions expected to take place this year, and the overall timeframe to implement the new core UK medical device regulation.
Additional Reading
You may be interested in the following resources:


{kind=link}
{kind=link}