
EU Guide: Importer Requirements under the MDR/IVDR

Team NB: New MDR Training Sessions Added for February/March 2024
November 16, 2023
Team NB Training Survey Results & EU Competent Authorities ‘MDR Call to Action’
November 28, 2023Page Last Updated: 4 January 2024
- Importer vs Distributor
- Importer’s Obligations (Article 13)
- Distributor’s Obligations (Article 14)
- Appointing an Importer: Is it Mandatory?
- Importer Agreement: Is it Required?
- Importer on the device labeling:
- Manufacturer & Importer: Can it be the Same Company?
- EC Rep & Importer: Can it be the Same Company?
- Direct Sales to End Users: Who is the Importer?
- Third-Party Logistics Provider: Are they the Importer?
- Pharmacy / Retailer: Are they the Importer?
- Legacy Devices: Importer/Distributor Requirements
Importer vs Distributor
The difference between an importer and distributor, is in the action.
An importer is the first person (natural or legal) that places an individual device from a third country, onto the Union market. Further explanation on the concept of ‘placing on the market’ can be found here.
The distributor is any person (natural or legal) in the supply chain, that is between the EU manufacturer/EU importer and the end user. i.e., they are involved after the device has already been placed on the Union market, and before it is supplied to the end user. The MDR/IVDR definition of a distributor states they are:
“any natural or legal person in the supply chain, other than the manufacturer or the importer, that makes a device available on the market, up until the point of putting into service”
Can an EU company be both an importer and distributor?
An EU company can be both an importer and distributor, depending on the situation. However, they cannot be both roles at the same time, for the same action. Either they have first placed the device onto the Union market from a third country (making them the importer), or they have not, and are only further supplying it after this action has occured (making them the distributor).
Further, each respective role is per individual device, not type of product. i.e., the action occurs per transaction that results in the change of ownership of a finished/manufactured device.
What happens to the distributor role, if you only have one EU customer who buys and resells your devices?
An importer can also ‘make a device available on the market, up until the point of putting into service‘. In other words, an importer can be your European customer who buys your products, first places them on the market, and then also further sells them to hospitals, clinics, pharmacies, etc. The importer would then be completing two actions: 1) ‘first placing the device on the Union market’ and 2) ‘making the device available on the market up until the point of putting into service’.
The Irish Competent Authority published a table which may also be useful (page 2):
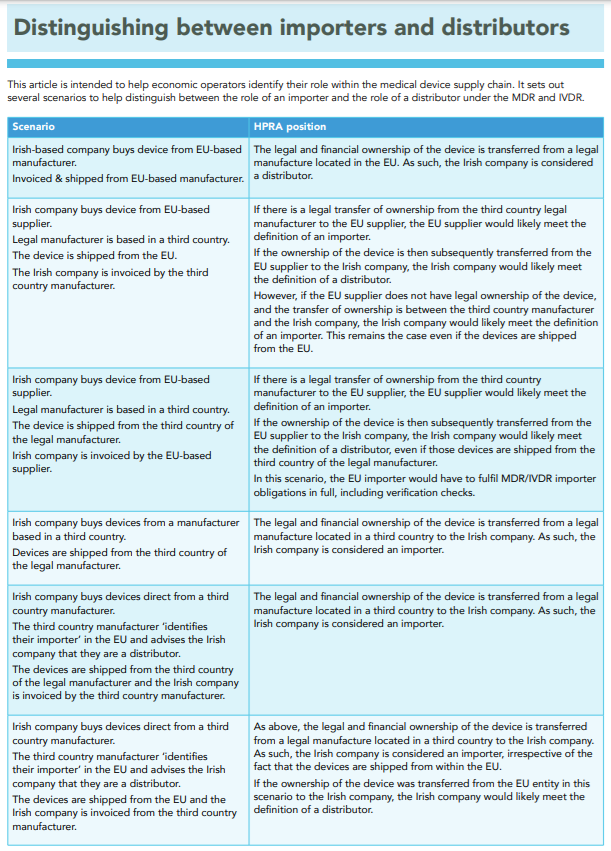
Importer’s Obligations (Article 13)
General Obligations
Per Article 13, the importer shall meet the following requirements (high-level):
- Review to ensure the device is properly CE Marked, including appointment of an EU Authorized Representative, drawing up a Declaration of Conformity, compliance with the labeling requirements and assignment of a UDI by the manufacturer
- Where an importer has reason to believe a device is not in conformity with the Regulation, they must not place those devices onto the market
- Identify themselves (name and address) on the device, on its packaging, or in a document accompanying the device (defined further below)
- Verify the device has been registered in EUDAMED*
- Register their own company in EUDAMED and link themselves with the manufacturers for whom they import
- While a device is under its responsibility, ensure storage and/or transport conditions are met
- Keep a register of complaints, of non-conforming devices and of recalls and withdrawals
- Forward any complaints from healthcare professionals, patients or users about suspected incidents to the manufacturer and the authorized representative
- Cooperate with the manufacturer, authorized representative and competent authorities for corrective actions, and any other measures to help eliminate and/or mitigate risks posed by the device
- Inform relevant parties if they believe: the device presents a serious risk, is a falsified device, or if a device they have placed onto the market is not in conformity with the Regulation
- Keep the declaration of conformity (and CE Certificate, if applicable) for the minimum period required under the Regulation
*EUDAMED is currently voluntary. For information on the most current registration deadlines, please read: EUDAMED Overview
Device traceability
Per Article 25 (MDR)/Article 22 (IVDR) ‘Identification within the supply chain’:
- Distributors and importers shall co-operate with manufacturers or authorised representatives to achieve an appropriate level of traceability of devices.
- Economic operators shall be able to identify the following to the competent authority, for the period referred to in Article 10(8):
- any economic operator to whom they have directly supplied a device;
- any economic operator who has directly supplied them with a device;
- any health institution or healthcare professional to which they have directly supplied a device.
This includes that:
“Economic operators shall store and keep, preferably by electronic means, the UDI of the devices which they have supplied or with which they have been supplied, if those devices belong to: (a) class III implantable devices; (b) the devices, categories or groups of devices determined by a measure referred to in point (a) of paragraph 11.
Other
There may be additional local Competent Authority requirements, such as obtaining an importer license.
For more information on EU registration requirements, please read: EU Registration Requirements Tool
Distributor’s Obligations (Article 14)
The distributor has similar requirements as the importer, with some differences.
Unlike the importer, distributor does not need to:
- Identify themselves with the device
- Register in EUDAMED
- Verify that the manufacturer has registered in EUDAMED
- Keep a register of complaints, of non-conforming devices and of recalls and withdrawals
Distributors are additionally responsible for:
- Ensuring the importer’s information has been applied per Article 13(3)
- While both the importer and distributor must confirm the device is CE Marked, labeled appropriately and that an EU Authorized Representative has been appointed, the distributor may “apply a sampling method that is representative of the devices supplied by that distributor”
Please see Article 14 for the full list of requirements.
Like importers, distributors may be subject to additional local Competent Authority requirements. For example, obtaining a distributor’s license or confirming which products they are placing on that individual member state’s market.
Appointing an Importer: Is it Mandatory?
While some manufacturers do intentionally ‘appoint’ an EU importer, ultimately the ‘appointment’ is out of their control. I.e., an importer is a role that occurs whether the manufacturer makes an active decision to ‘designate’ that importer or not.
Per the MDR/IVDR, any person (natural or legal) who first places a device from a third country onto the Union market, becomes the importer. Therefore, it is not a question of: is an importer mandatory or not. An importer is a de facto role that occurs when that action is taken. This is whether either party (manufacturer or importer) wanted or intended it.
For example, let us say you formally ‘appoint’ a company to act as your EU importer. You then implement processes to ensure your employees do not unintentionally sell/transfer ownership of products to any other EU company/importer. However, your distributor in the UK sells your products to their EU customer. That EU customer now becomes the EU importer of those products, as they are the entity first placing a product from a third country onto the Union market. This is despite your separate EU importer ‘appointment’ and internal processes. That other EU customer must now meet the importer obligations, including identifying themselves with the device.
MDCG 2021-27 Rev.1 (page 6) addresses how an importer should handle a situation where the devices are already labeled with another importer’s information.
“What should an importer do in the case where an individual device already mentions another importer’s details on its packaging?”
“In the unusual case where details of another importer already appear on the packaging of an individual device (for example, the individual device has been exported and then reintroduced to the Union market), the importer should verify if the individual device has previously been placed on the Union market. This may be done by contacting the manufacturer. The importer should replace any previous importer details with their own, if having investigated the issue, they determine themselves to be the correct importer. The label with the previous details will be void.”
So, while you may ‘appoint’ an importer, if a different EU entity first places your devices onto the Union market, they become the importer for those products. They must add their own information to the devices and meet all the other obligations outlined in the regulation.
Importer Agreement: Is it Required?
The MDR/IVDR does not explicitly require a formal contract between the manufacturer and importer (or distributor). This is unlike the manufacturer and its authorized representative: written and accepted mandate required. And the manufacturer and its Notified Body: signed agreement required.
Manufacturers may choose to put an agreement in place with their importer for other reasons. For example, to cover commercial issues, such as sales and payment terms. Or, as a part of its Quality Management System, related to supplier controls. And in that agreement, the manufacturer may stipulate that importers must meet their obligations under MDR/IVDR, such as ensuring the importer’s information is supplied with the product, device traceability, forwarding complaints, etc. The agreement may also allow the manufacturer to cancel the agreement if the importer does not meet its regulatory obligations.
Whether an agreement is in place or not, importers/distributors are legally obligated to meet their minimum requirements under the MDR/IVDR.
Importer on the device labeling:
Who is responsible for identifying the importer (i.e., ‘labeling’)?
It is the importer’s obligation to ensure that their information is appropriately supplied with the device.
Per MDCG 2021-27 Rev.1 (page 6):
“Who is responsible for indicating the importer on the device, its packaging or accompanying documentation?”
“Importers are responsible for including their information on the device, its packaging or in accompanying documentation in accordance with Article 13(3) of the Regulations. The importer may add this information themselves or sub-contract this task to the manufacturer, however the importer remains responsible for the correct execution of the obligation, regardless of the means chosen.”
This means the manufacturer may ship products without the importer information applied. The importer would then be required to include their details prior to placing the products onto the market.
Per MDCG 2021-27 Rev.1:
“Whilst the inclusion of the importer’s details before the device has physically entered the Union is not mandatory, the importer’s details must be included on the device (or on its packaging, or in a document accompanying the device) when the device is placed on the Union market (i.e. the first making available). The absence of the importer’s details at customs control should therefore not be considered as a non-compliance with the Regulations.”
Therefore, while the importer and manufacturer can mutually agree that the manufacturer will add this information to the labeling, it is ultimately the importer’s obligation to ensure that the information is correctly applied.
What is a ‘document accompanying the device’?
MDR/IVDR Article 13(3) requires that the importer’s details be provided on the product, the packaging or in a document accompanying the device.
MDCG 2021-27 Rev.1 (page 6) states that the ‘accompanying documentation’ with the importer’s information may be separate from, or affixed to, the individual device. It can be a sticker affixed to the label or a separate leaflet. The main point is that it must accompany the individual device throughout the supply chain and reach the end user. As best practice, the importer should consider providing the accompanying documentation with the smallest package that can be purchased by the end user.
The accompanying documentation should allow the importer to be located and contacted, and allow healthcare professionals, patients, and users to report any suspected incidents to the importer.
Where can I get the importer symbol?
The importer symbol is available in EN ISO 15223-1:2021, which has been harmonized under the MDR and IVDR. Therefore, this symbol may be used on labeling without having to be further defined.
Companies may purchase a copy of EN ISO 15223-1:2021 for the full list of European harmonized symbols.
Otherwise, there are multiple places to obtain a copy of the importer symbol, such as MedTech Europe’s ‘Use of Symbols to Indicate Compliance with the MDR’.
For more information, please read: List of MDR/IVDR Harmonized Standards & Common Specifications
Is the distributor required on the labeling?
No, the distributor does not need to identify themselves with the device. Instead, their responsibility is to ensure that the importer has been properly identified with the device.
That said, the distributor may voluntarily be added to the labeling. This may help ensure they are correctly differentiated from the importer.
If the distributor information is added, there is a harmonized symbol in EN ISO 15223-1:2021 that may be used.
Manufacturer & Importer: Can it be the Same Company?
An importer is a person that places a device from a third-country onto the Union market. An EU manufacturer will not have an importer because they 1) are not from a third country and 2) will (presumably) be the first person to place the device on the Union market.
Therefore, this question is answered more specifically as: Can a non-EU manufacturer set up a subsidiary to act as the EU importer?
The answer to that is “yes”. There is nothing in the Regulation that prevents it, and the MDCG guidance on importers and distributors does not address this topic. Further, there are many examples of companies who have set-up this infrastructure, as seen in EUDAMED.
That said, companies may want to consider the original intent of the MDR/IVDR when doing so. The importer is tasked with ensuring that the manufacturer has met the conformity assessment requirements. Further, the importer should report the manufacturer if they have any “reason to believe that the device presents a serious risk or is a falsified device”. There is an obvious conflict of interest, when the company tasked with this obligation, is a subsidiary of the company they must report.
If manufacturers opt to set-up an EU subsidiary, they may want to consider how to properly address the responsibilities between the two economic operators. For example, if the importer is inspected by its national competent authority, can it defend that the checks-and-balances are not compromised? Is the importer sufficiently enabled to meet its obligations, including to report a non-compliance against its own parent company?
EC Rep & Importer: Can it be the Same Company?
Yes, the EU Authorized Representative (EC Rep) can act in both capacities – as the EC Rep and the importer.
In this case, that entity must meet the obligations of each respective role: MDR/IVDR Article 11, Authorized Representative and MDR/IVDR Article 13, General obligations of importers. And further, be able to demonstrate how they are meeting the requirements of each individual role.
Direct Sales to End Users: Who is the Importer?
For example, if a software product (SaMD) or physical device is purchased online directly by the end user, who is the EU importer in this case?
Unfortunately, MDCG 2021-27 Rev.1 does not address direct sales. Therefore, until the MDCG produces a better answer, we have to go to other sources, such as the Blue Guide and related legislation.
The short conclusion is that the end user who purchased the device, likely does not take on the obligations of the importer.
Read below for full rationale.
From the perspective of direct sales to an end user, Section 3.8 of the Blue Guide states:
“The end user is any natural or legal person residing or established in the Union, to whom a product has been made available either as a consumer outside of any trade, business, craft or profession or as a professional end user in the course of its industrial or professional activities.
Union harmonisation legislation does not create obligations for the end-users of the products in their scope.
This is the case even when there are no responsible economic operators present within the EU (for example, in the context of products sold online and for which an economic operator under Article 4 of Regulation (EU) 2019/1020* is not required. The term thus covers both professional users and consumers.”
Regulation (EU) 2019/1020, cited above, indicates that an EU Economic Operator may by default assume additional responsibilities, in the absence of other applicable EU economic operators. However, it also states that Article 4 is only applicable to certain legislation, of which the MDR/IVDR are not included.
Therefore, considering:
- the Blue Guide’s statement that end-users (both consumers and professionals) are not subject to legislative obligations, e.g., importer requirements under the MDR/IVDR, and
- lack of clear guidance from the MDCG
It could be considered that there is no EU importer (as defined by the MDR/IVDR) in this situation.
Therefore, manufacturers could:
- Draft a rationale, citing the Blue Guide, as to why there is no EU importer for direct sales to end users. This could then be presented to the notified body or competent authority in discussions regarding the supply chain and economic operator roles, if/when applicable.
- Companies that have a high volume of direct sales (whether the product is physically shipped or remotely downloaded) could hire a third-party company to act as a dedicated importer for these products. This may help remove any uncertainties, although may add to the overall cost to market the device in the EU.
The EU Commission themselves have created this uncertainty by not answering this question in any MDR/IVDR specific guidance documents. Therefore, it should be reasonable to conclude that manufacturers will create a rationale based on the materials that are currently available.
*General EU legislation on market surveillance and compliance of products.
Third-Party Logistics Provider: Are they the Importer?
Whether or not the Third-Party Logistics Provider (3PL) is the importer, depends on what they have been contracted to do.
Per MDCG 2021-27 Rev. 1 (page 6):
“Some 3PL companies which provide transportation services or hold devices on a consignment basis only (i.e. where devices are held at a site by the 3PL, but the 3PL does not have legal ownership of those devices), may not be considered an importer provided there is a clearly defined agreement between both parties setting out the responsibilities of each party.
The importer is the natural or legal person meeting the definition of Article 2(33) MDR/Article 2(26) IVDR, with ownership, possession or any other property right over the device.
Although transportation or storage activities may be subcontracted outside of the importer’s organisation, the importer retains responsibility over storage and transport conditions and as such, must ensure the sub-contractor’s conditions do not jeopardise compliance with the general safety and performance requirement of Annex I of the Regulations (see Article 13(5) of the Regulations).”
Pharmacy / Retailer: Are they the Importer?
Per MDCG 2021-27 Rev.1, yes, it is possible that individual shops, pharmacies, retailers and other persons can be importers or distributors under the MDR/IVDR.
“A distributor is any natural or legal person in the supply chain, other than the manufacturer or the importer, that makes a device available on the market, up until the point of putting into service (see Article 2(34) MDR/Article 2(27) IVDR). As such, individual shops, pharmacies or retailers or other natural or legal persons meeting this definition, are considered distributors.
For example, a community pharmacy, an individual shop, retailer or other person, which buys and then sells type II medical face masks to customers (whether online or physically), such as other shops or companies or private individuals, are considered to supply medical devices and thereby fall within the definition of a distributor. These entities will be expected to comply with Article 14 of the Regulations and any applicable national registration requirements.
Furthermore, these operators will assume the role and responsibilities of an importer if they obtain the device directly from a non-EU based manufacturer or distributor and are expected to comply with Article 13 of the Regulations.”
Legacy Devices: Importer/Distributor Requirements
Per MDCG 2021-25 (application of MDR requirements to ‘legacy’ devices) and MDCG 2022-8 (application of IVDR requirements to ‘legacy’ devices) , some aspects of the MDR/IVDR apply to importers and distributors.
Particularly, those outlined in the MDR/IVDR transition articles, e.g., post-market surveillance, market surveillance, vigilance and registration of economic operators and devices, which apply to all legacy devices.
Please see below the table of Article 13 importer requirements. Each section is noted applicable/not applicable for MDD/AIMDD/IVDD legacy devices per the above MDCG guidance documents.
| Applicable Per MDCG 2021-25 (MDR) and 2022-8 (IVDR) | Description |
| 13(1) 13(2)(a)-(d) | NOT APPLICABLE – obligation to verify (and document such verification) the device holds valid conformity assessment. This includes that the Declaration of Conformity has been drawn up, the manufacturer and EU authorized representative (if applicable) are identified, the device is appropriately labeled, and that UDI has been assigned in accordance with Article 27 (where applicable). |
| 13(2), second subparagraph | APPLICABLE – Where an importer considers or has reason to believe that a device is not in conformity with the requirements of this Regulation, it shall not place the device on the market until it has been brought into conformity and shall inform the manufacturer and the manufacturer’s authorised representative. Where the importer considers or has reason to believe that the device presents a serious risk or is a falsified device, it shall also inform the competent authority of the Member State in which the importer is established. |
| 13(3) | NOT APPLICABLE – obligation to add the importer’s details to the device. |
| 13(4) | APPLICABLE – Importers shall verify that the device is registered in the electronic system in accordance with Article 29. Importers shall add their details to the registration in accordance with Article 31. |
| 13(5) | NOT APPLICABLE – obligation to ensure storage or transport conditions do not jeopardise its compliance with the general safety and performance requirements under Annex I of the MDR/IVDR. |
| 13(6) | APPLICABLE – Importers shall keep a register of complaints, of non-conforming devices and of recalls and withdrawals, and provide the manufacturer, authorised representative and distributors with any information requested by them, in order to allow them to investigate complaints. |
| 13(7) | APPLICABLE – Importers who consider or have reason to believe that a device which they have placed on the market is not in conformity with this Regulation shall immediately inform the manufacturer and its authorised representative. Importers shall co-operate with the manufacturer, the manufacturer’s authorised representative and the competent authorities to ensure that the necessary corrective action to bring that device into conformity, to withdraw or recall it is taken. Where the device presents a serious risk, they shall also immediately inform the competent authorities of the Member States in which they made the device available and, if applicable, the notified body that issued a certificate in accordance with Article 56 for the device in question, giving details, in particular, of the non-compliance and of any corrective action taken. |
| 13(8) | APPLICABLE – Importers who have received complaints or reports from healthcare professionals, patients or users about suspected incidents related to a device which they have placed on the market shall immediately forward this information to the manufacturer and its authorised representative. |
| 13(9) | NOT APPLICABLE – obligation to keep a copy of the EU declaration of conformity and copy of any relevant certificate (if applicable) for the minimum time set forth under the MDR/IVDR. |
| 13(10) | APPLICABLE – Importers shall cooperate with competent authorities, at the latters’ request, on any action taken to eliminate or, if that is not possible, mitigate the risks posed by devices which they have placed on the market. Importers, upon request by a competent authority of the Member State in which the importer has its registered place of business, shall provide samples of the device free of charge or, where that is impracticable, grant access to the device. |


{kind=link}
{kind=link}