
EU Guide: Basic UDI-DI

Certificates of Free Sales for Medical Devices: Europe, UK and Switzerland
January 3, 2022
UK Steps to Market
January 4, 2022While Basic UDI-DI was created to help streamline device identification, it continues to be a confusing concept to many. Even though MDCG guidance document 2018-1 Rev.4 has been published on Basic UDI-DI, it still leaves a lot unanswered. Below we help clarify some of the unknowns.
- UDI, UDI-DI and UDI-PI Primer
- What is Basic UDI-DI?
- Example: multiple devices linked to a Basic UDI-DI
- Is Basic UDI-DI needed if I only have a one device?
- Is Basic UDI-DI needed for Systems, Procedure Packs & Kits?
- Is Basic UDI-DI needed for ‘legacy’ devices?
- Is Basic UDI-DI needed for custom-made devices?
- Where will the Basic UDI-DI be listed?
- Is the Basic UDI-DI required on the labeling?
- How do you obtain a Basic UDI-DI?
- What does a Basic UDI-DI look like?
- Basic UDI-DI Recap
UDI, UDI-DI and UDI-PI Primer
UDI (Unique Device Identification) stated on its own generally refers to the entire system for device traceability. This includes the Basic UDI-DI (for Europe & Switzerland only), the UDI-DI and UDI-PI. As well as a host of other considerations, such as the UDI carrier (e.g., bar code).
UDI-DI refers to the ‘Device Identifier’ for a specific product. The UDI-DI is a fixed numeric or alphanumeric code, and each UDI-DI is associated with one individual product model/version. The UDI-DI contains the following within it: device name, model, reference or catalogue number, clinical size, risk class, whether it is labelled as a single use device, critical warnings or contra-indications and more.
UDI-PI refers to the ‘Production Identifier’ for the unit of device production. The UDI-PI is also a numeric or alphanumeric code; however, it is not a fixed code. The UDI-PI is variable because it is applicable to non-fixed data such as the serial number, lot number, software identification, manufacture date and/or expiration date.
The MDR/IVDR makes implementation of a UDI system mandatory in Europe. UDI was not a requirement under the old Directives.
What is Basic UDI-DI?
A Basic UDI-DI code is intended to link together all devices with the ‘same intended purpose, risk class and essential design and manufacturing characteristics’.
A UDI-DI, on the other hand, is the identification code for a specific product model/version. The UDI-DI links up into the Basic UDI-DI.
Think of the Basic UDI-DI as the “parent” identifier, and underneath it are all the UDI-DIs within a common device “family”.
The EU Commission’s sample structure is helpful to envision this.
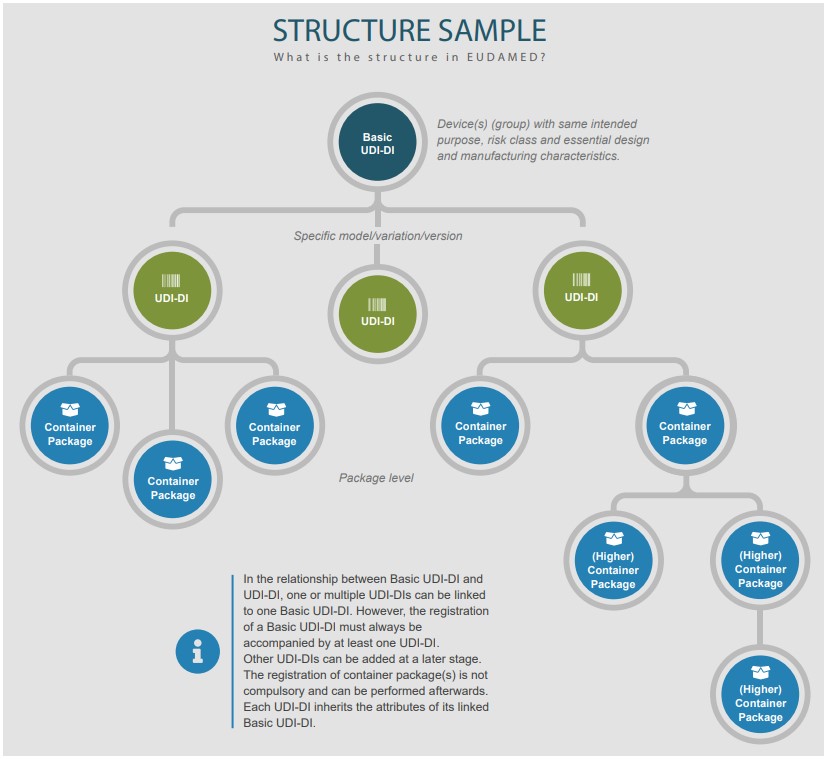
Example: multiple devices linked to a Basic UDI-DI
Let us say you manufacturer finger pulse oximeters.
You have a group of product models that share the same intended purpose, classification and essential design and manufacturing characteristics, and you have therefore included these product models within the same Technical Documentation File (TDF), and you have issued a single Declaration of Conformity (DoC), identifying all the models within it. You title the TDF ‘Finger Pulse Oximeters’. In this case, you may have one Basic UDI-DI for all the device models/variations within that ‘group of devices’. Each of the models have their own unique UDI-DIs. And all of those devices/UDI-DIs would be linked to that single Basic UDI-DI.
Is Basic UDI-DI needed if I only have a one device?
Yes. Basic UDI-DI is still necessary even if you only have a single device/device model and do not have multiple products within a common device “family”. In this case, you will have one Basic UDI-DI and one UDI-DI.
If you later add a new trade name, version or model, or vary characteristics (e.g., one version single-use, another version reusable), then a new UDI-DI is required for that differing device. The new UDI-DI generated for that product will feed up into the already existing Basic UDI-DI. Thus, the original and new UDI-DIs become connected under the same Basic UDI-DI.
Is Basic UDI-DI needed for Systems, Procedure Packs & Kits?
Yes. A Basic UDI-DI is required for Systems, Procedure Packs and Kits if they are marketed under the MDR/IVDR.
Each is defined as the following in the EU Commission’s EUDAMED Infographic Categorization of Devices:
‘System’ … means a combination of products, either packaged together or not, which are intended to be inter-connected or combined to achieve a specific medical purpose.
‘Procedure pack’ … means a combination of products packaged together and placed on the market with the purpose of being used for a specific medical purpose.
‘Kit’ … means a set of components that are packaged together and intended to be used to perform a specific in vitro diagnostic examination, or a part thereof.
If the System, Procedure Pack, IVD Kit is marketed under the old Directives, then a EUDAMED DI / EUDAMED ID may be required instead. Please see the below section on legacy devices for more information on this topic.
Is Basic UDI-DI needed for ‘legacy’ devices?
A Basic UDI-DI is not required for devices CE marked under the old Directives – it is only mandatory for MDR/IVDR CE marked devices.
However, that doesn’t mean that ‘legacy’ device manufacturers are completely exempt from this requirement. For the purposes of registering in EUDAMED, legacy devices will require a EUDAMED DI, which will perform a similar function as the Basic UDI-DI.
Where a legacy device manufacturer has a UDI system in place and UDI-DI applied for its devices, EUDAMED will auto-generate a EUDAMED DI when they register. Where a legacy device manufacturer does not yet have a UDI system in place, because UDI is not mandatory under the old Directives, then one will need to be created independent from a UDI-DI. This is called the EUDAMED ID.
The below is from the EU Commission’s User Guide:
EUDAMED DI: The EUDAMED DI corresponds to the Basic UDI-DI. It can either be entirely generated by EUDAMED if a UDI-DI has already been assigned to the legacy device, or the DI code can be partly assigned by the manufacturer (EUDAMED is the issuing entity for a EUDAMED DI)
EUDAMED ID: The EUDAMED ID corresponds to the UDI-DI. In case an UDI-DI has not already been assigned, the EUDAMED ID will always be automatically and fully generated by EUDAMED from the EUDAMED DI.
It can be somewhat of a confusing concept. But essentially, legacy devices will either have a EUDAMED DI or a EUDAMED ID, depending on if the manufacturer has a UDI system in place yet or not. Both the EUDAMED DI and EUDAMED ID are auto-created within the EUDAMED database.
This is different than the process for a Basic UDI-DI, where the manufacturer must procure the Basic UDI-DI outside of EUDAMED and then enter into EUDAMED.
Further, the EUDAMED DI / EUDAMED ID is not required to be listed in the device’s technical file, Declaration of Conformity, nor added to your MDD/AIMDD/IVDD CE Certificates. However, manufacturers may include the EUDAMED DI / EUDAMED ID to some technical documentation or procedures, as necessary for device traceability.
A final note on this topic is that EUDAMED is currently voluntary. Read more about registration requirements and timing on our EUDAMED Basics page.
Is Basic UDI-DI needed for custom-made devices?
No. Custom-made devices are exempt from Basic UDI-DI.
Custom-made devices (CMD) are not subject to UDI requirements as a whole, per MDR Article 27(1):
“The Unique Device Identification system (‘UDI system’) described in Part C of Annex VI shall allow the identification and facilitate the traceability of devices, other than custom-made and investigational devices, and shall consist of the following…”
Article 29(1) further states:
“Before placing a device, other than a custom-made device, on the market, the manufacturer shall, in accordance with the rules of the issuing entity referred to in Article 27(2), assign a Basic UDI-DI as defined in Part C of Annex VI to the device and shall provide it to the UDI database together with the other core data elements referred to in Part B of Annex VI related to that device.”
The MDCG guidance document 2021-3 also states:
“CMD manufacturers are exempt from device UDI registration, assignment and labelling requirements. As such, and although they must appoint a person responsible for regulatory compliance (PRRC) in accordance with Article 15 of the MDR, they are not required to register these persons in EUDAMED.”
Where will the Basic UDI-DI be listed?
The Basic UDI-DI must appear in the following:
- Technical Documentation File (Annex II, Section 1.1(b))
- Declaration of Conformity (Annex IV)
- CE Certificate* (Annex XII, Section 4(a))
- Summary of Safety and Clinical Performance (Art. 32 for implantable & Class III)
- Certificates of Free Sale** (Art. 60)
- EUDAMED device registration (Art. 29)
*If you leverage your CE Certificate for an abridged review in a country like Australia or Singapore, those regulatory authorities will be able to identify all associated device models/versions since the Basic UDI-DI is listed on the certificate.
**Certificates of Free Sale are often used for registration in markets such as Saudi Arabia or the Philippines.
Is the Basic UDI-DI required on the labeling?
No. The Basic UDI-DI is not required on the device labeling. It is only visibly listed, in human readable format, on documentation such as the Technical Documentation File, Declaration of Conformity, CE Certificate and Certificates of Free Sale.
That said, it is part of the Unique Device Identification (UDI) system, and therefore will be contained within the UDI-DI. The UDI carrier (e.g., barcode) is required on the device labeling, and in this sense, the Basic UDI-DI is on the labeling. When the UDI carrier is scanned, the Basic UDI-DI will be identifiable.
There is a transition period for the UDI carrier to be appear on the device labeling:
MDR, Art. 123(f):
- For implantable devices and for class III devices Article 27(4) shall apply from 26 May 2021
- For class IIa and class IIb devices Article 27(4) shall apply from 26 May 2023
- For class I devices Article 27(4) shall apply from 26 May 2025
MDR, Art. 123(g):
…with regard to reusable devices that are required to bear the UDI carrier on the device itself, Article 27(4) shall apply to:
- implantable devices and class III devices from 26 May 2023
- class IIa and class IIb devices from 26 May 2025
- class I devices from 26 May 2027
IVDR, Art. 113(e):
- For class D devices, Article 24(4) shall apply from 26 May 2023
- For class B and class C devices Article 24(4) shall apply from 26 May 2025
- For class A devices Article 24(4) shall apply from 26 May 2027
How do you obtain a Basic UDI-DI?
The Basic UDI-DI must be generated by your UDI issuing agency. You as the manufacturer do not make up this code on your own. In fact, the validity of the Basic UDI-DI you enter into the EUDAMED database will be verified before you can proceed to the next step.
Many manufacturers already have UDI-DI codes because they are selling in a country that requires it, such as the USA. In this case, all you need to do is obtain one additional code from your issuing agency – the Basic UDI-DI.
The caveat is that the issuing agency must be one of the following organizations. Only these four are recognized by the EU Commission:
- GS1
- HIBCC
- ICCBBA
- IFA GmbH
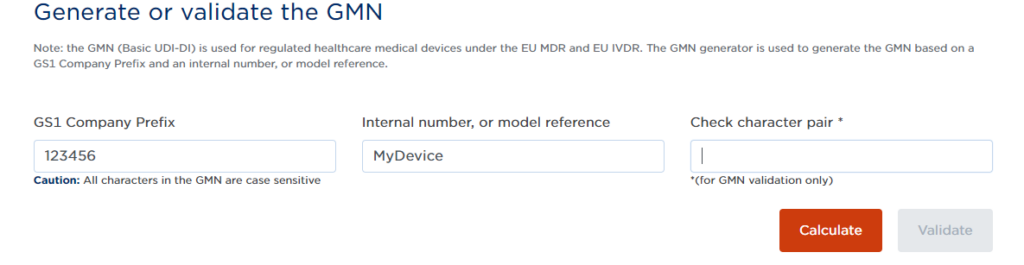
Some of the issuing entities have an online Basic UDI-DI generator for their customers:
You can use these to generate your Basic UDI-DI. For example, for GS1 you would enter your GS1 Company Prefix + Internal Reference, and then click “calculate”. The site will then generate your Basic UDI-DI for that product family.
What does a Basic UDI-DI look like?
Per MDCG Guidance document 2019-1, at minimum, a Basic UDI-DI must have:
- Format that is as close as possible to the UDI-DI
- Maximum of 25 characters
- Check/digit character, based on an algorithm defined by the issuing entity
As a result, the Basic UDI-DI’s structure will differ depending on the which UDI issuing entity you select. Below are example Basic UDI-DIs. Note that the mandatory ‘check/digit’ is in bold for reference.
- HIBCC: ++A999MODELIDENTIFIER11S8
- ICCBBA: A9990ABC123T048008
- IFA: PP12345ABCD.12345678.9004
- GS1: no specific example given, however does outline the structure of the Basic UDI-DI, including how the check character appears for GS1 customers – “two uppercase alpha and numeric characters”, e.g., 1234567891234XR
Basic UDI-DI Recap
| Yes/No | Requirements |
|---|---|
| Yes | Is required for MDR/IVDR CE marked devices |
| No | Is required for MDD/AIMDD/IVDD CE marked legacy devices |
| Yes | Is a top-level ‘parent’ UDI, under which a group of like devices are bundled |
| No | Is required per device model within the device family |
| No | Is required for custom-made devices |
| Yes | Must be issued by one of the four EU Commission recognized UDI issuing agencies |
| No | Is on the device labeling and/or any other marketing materials |
| Yes | ls required on the following: – Technical Documentation File – Declaration of Conformity – CE Certificate – Summary of Safety and Clinical Performance (implantable & Class III) – Certificates of Free Sale – EUDAMED device registration |


{kind=link}
{kind=link}